Research Focus
We take an interdisciplinary approach that combines proteomics technology with biological inquiries
RNA protein correlations
Machine learning and deep learning methods to predict proteins from mRNA
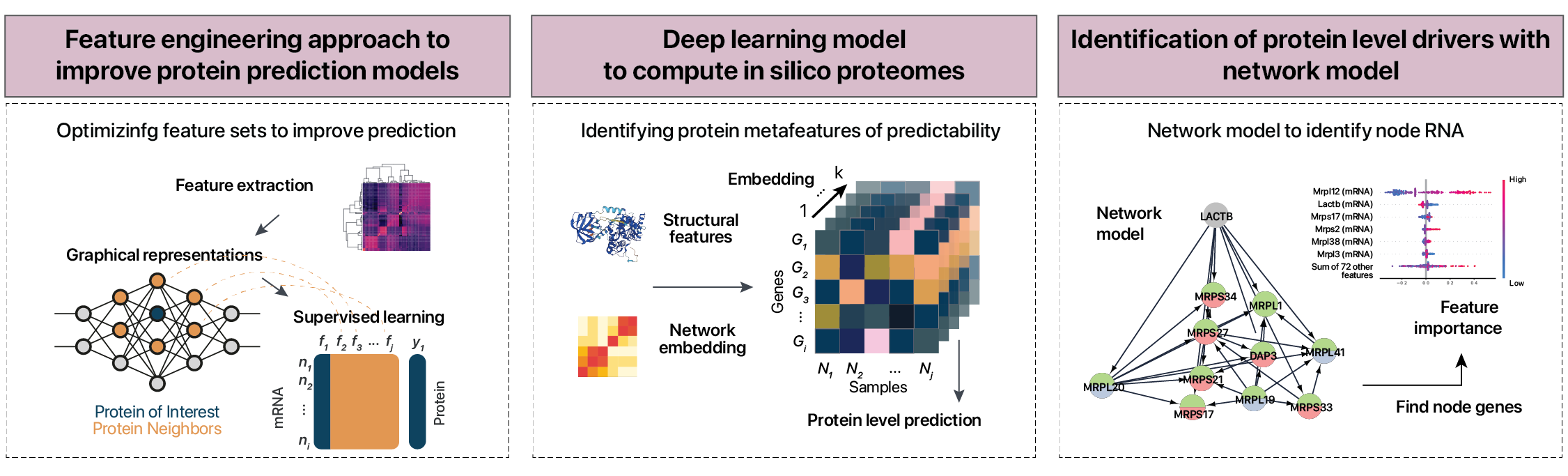
The levels of mRNA in the cell is very commonly measured as a readout of genome output. In the last few years, there has been an increasing realization among scientists of the imperfect correlation between mRNA and protein levels. Although a robust correlation exists between protein and mRNA across different genes (i.e., highly abundant mRNAs tend to produce highly abundant proteins within a cell), there is often only modest correlation between the fold-change of a particular mRNA and the corresponding change of the protein it produces upon perturbation or disease. Our lab is interested in using machine learning and deep learning tools to improve the prediction of protein levels from mRNA data. Some projects include:
Protein prediction models
This unexplained protein variance may be attributed, in part, to the many post-transcriptional and post-translational regulatory mechanisms that modulate protein concentration in the cell. For example, large multi-protein complexes are known to buffer gene copy number changes, because the subunits that are surplus to the stoichiometry of assembly cannot fold properly, and become degraded by the cell. Other mechanisms such as gene-specific translation rates of mRNA also contribute to the biophysical and biological barriers that uncouple the mRNA and protein layers of biological information. These mechanisms are usually not directly revealed from transcriptomics data that aim tp measure differentially expressed mRNA. As a result, there is a need to examine critically mRNA- protein relationship, and the refinement of methods to predict protein changes from mRNA data is a relevant goal to many fields of biomedical research. Our lab aims to apply machine learning methods to predict the across-sample protein variance from RNA-seq data. Using this approach, we are discovering the drivers behind huge gene-wise differences in predictability. For instance, up to 1/3 of proteins are poorly predicted by mRNA. Many proteins show very poor correlation with their cognate mRNA but instead a strong correlation with another transcript, which are usually but not always their known protein-protein interaction partners. The data suggests degradation of supernumerary interactors is a driver of protein levels. Although this has been known for some time for some large multi-subunit complexes like the ribosome, our research suggests that this phenomenon is in fact very widespread in the cell and affects many small stable complexes incl. propionyl-CoA carboxylase, mito. calcium uniporter, calcineurin, etc.
Improving predictions by feature selection and deep learning
In parallel, we are exploring the use of prior feature selection and engineering to improve predictions. For instance, pre-selecting the mRNA some genes that may predict the levels of a proteins not only improved protein predictions, but may also help find new protein-level driver genes. Using a directed graph model, we predict that the LACTB mRNA may have an outsized effect on mitochondrial ribosome protein abundance. Some of our recent findings can be found in our paper “widespread post-transcriptional regulation of protein abundance by interacting partners” on PLoS Computational Biology. Ongoing work is applying deep learning and explainable AI to further explore how cells regulate proteome profiles independently from mRNA.
Application to aging research
Understanding the molecular mechanisms of aging hearts and skeletal muscles is an important component in the quest to mitigate and prevent prevalent morbidities in an aging world. RNA-protein correlation may be an important part of the aging process, as senescent cells tend to have further decreased concordance in gene expression level at the RNA vs. protein levels, suggesting gene regulatory programs in aged cells may not be faithfully transmitted to produce a healthy proteome. In prior work, we derived a computational workflow that uses how well RNA predicts protein to help understand aging proteome regulation. We found that only about half of the RNA targets in an aging comparison study might reasonably predict protein level changes. These results help highlight a subset of potential targets for follow-up studies that may help inform aging mechanism
Team Members

Edward Lau
PI
Calvin Voong
POSTDOCRelated Publications
Protein prediction models support widespread post-transcriptional regulation of protein abundance by interacting partners.
Srivastava, Himangi , Lippincott, Michael J , Currie, Jordan , …
PLoS Comput Biol (2022)Transcriptome features of striated muscle aging and predictability of protein level changes.
Han, Yu , Li, Lauren Z , Kastury, Nikhitha L …
Mol Omics (2021)Last updated: January 27, 2026